Detailed walk-through of de novo CIDER (dnCIDER) on pancreas data
Source:vignettes/dnCIDER.Rmd
dnCIDER.RmdIntroduction
This vignette performs dnCIDER on a cross-species pancreas dataset. It is aimed to show the underneath structure of dnCIDER compared to the other high level vignette.
Load example data
The example data can be downloaded from https://figshare.com/s/d5474749ca8c711cc205.
Pancreatic cell data\(^1\) contain cells from human (8241 cells) and mouse (1886 cells).
load("../data/pancreas_counts.RData") # count matrix
load("../data/pancreas_meta.RData") # meta data/cell information
seu <- CreateSeuratObject(counts = pancreas_counts, meta.data = pancreas_meta)
table(seu$Batch)
#>
#> human mouse
#> 8241 1886Perform initial clustering
seu_list <- Seurat::SplitObject(seu, split.by = "Batch")
seu_list <- mclapply(seu_list, function(x) {
x <- NormalizeData(x, normalization.method = "LogNormalize",
scale.factor = 10000, verbose = FALSE)
x <- FindVariableFeatures(x, selection.method = "vst",
nfeatures = 2000, verbose = FALSE)
x <- ScaleData(x, verbose = FALSE, vars.to.regress = "Sample")
x <- RunPCA(x, features = VariableFeatures(object = x), verbose = FALSE)
x <- FindNeighbors(x, dims = 1:15, verbose = FALSE)
x <- FindClusters(x, resolution = 0.6, verbose = FALSE)
return(x)
})| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| acinar | 0 | 0 | 823 | 0 | 0 | 0 | 1 | 1 | 2 | 102 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 |
| activated_stellate | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 269 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 4 | 0 |
| alpha | 1067 | 3 | 0 | 0 | 0 | 665 | 0 | 0 | 0 | 0 | 251 | 243 | 1 | 6 | 0 | 0 | 1 | 0 | 4 |
| b_cell | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| beta | 1 | 886 | 2 | 812 | 740 | 1 | 0 | 0 | 0 | 0 | 2 | 0 | 3 | 2 | 1 | 0 | 4 | 0 | 1 |
| delta | 0 | 3 | 2 | 1 | 3 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 215 | 1 | 216 | 0 | 148 | 0 | 2 |
| ductal | 1 | 1 | 6 | 0 | 0 | 0 | 469 | 386 | 0 | 169 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| endothelial | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 212 | 0 | 1 | 0 |
| epsilon | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 8 | 0 | 0 | 1 | 0 | 0 |
| gamma | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 3 | 210 | 0 | 0 | 25 | 0 | 0 |
| immune_other | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| macrophage | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 39 |
| mast | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 25 |
| quiescent_stellate | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 153 | 0 |
| schwann | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 10 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| t_cell | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 |
seu_list <- mclapply(seu_list, RunTSNE, dims = 1:15)
p1 <- scatterPlot(seu_list[[1]], "tsne", colour.by = "seurat_clusters")
p2 <- scatterPlot(seu_list[[2]], "tsne", colour.by = "seurat_clusters")
cowplot::plot_grid(p1,p2)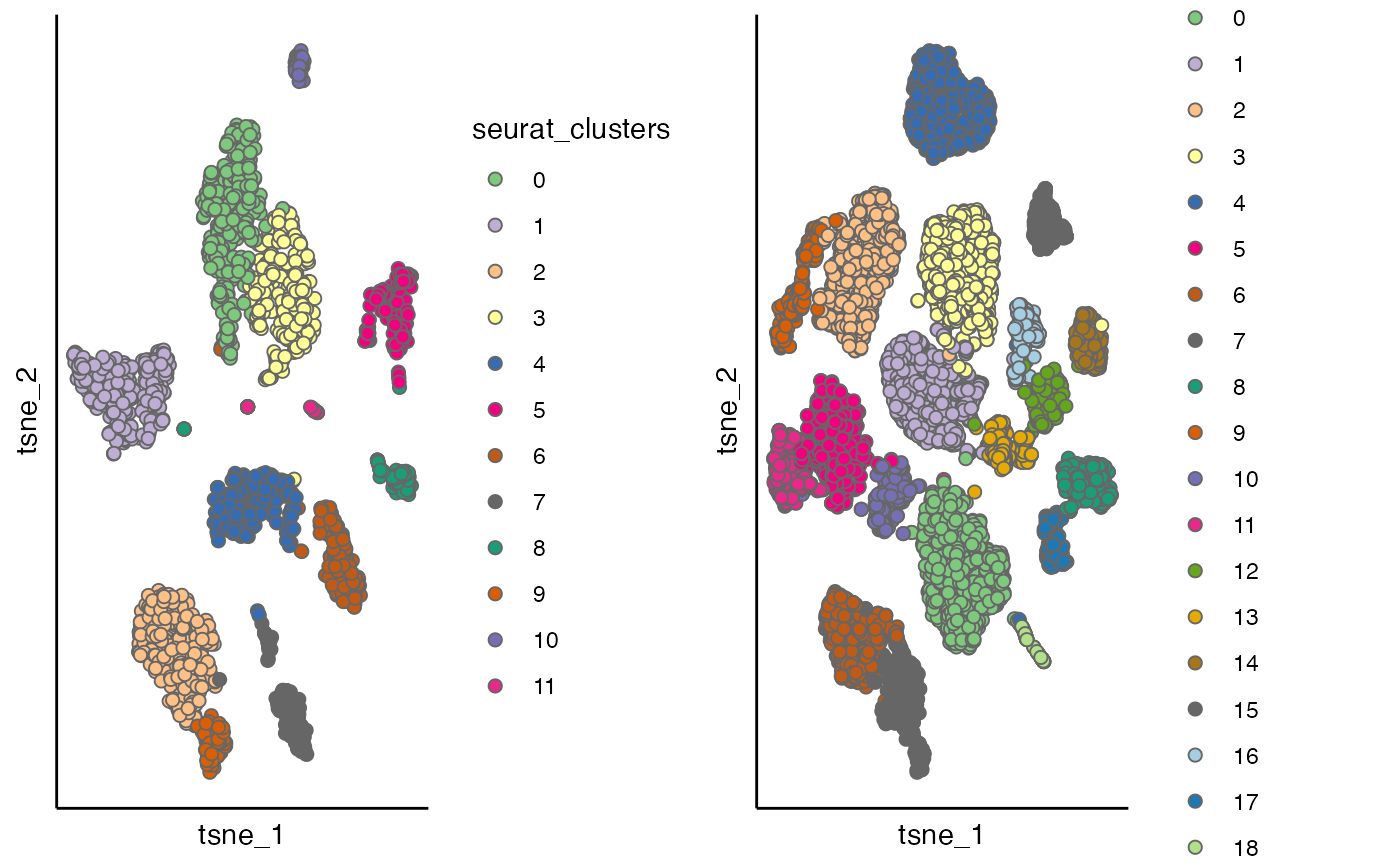
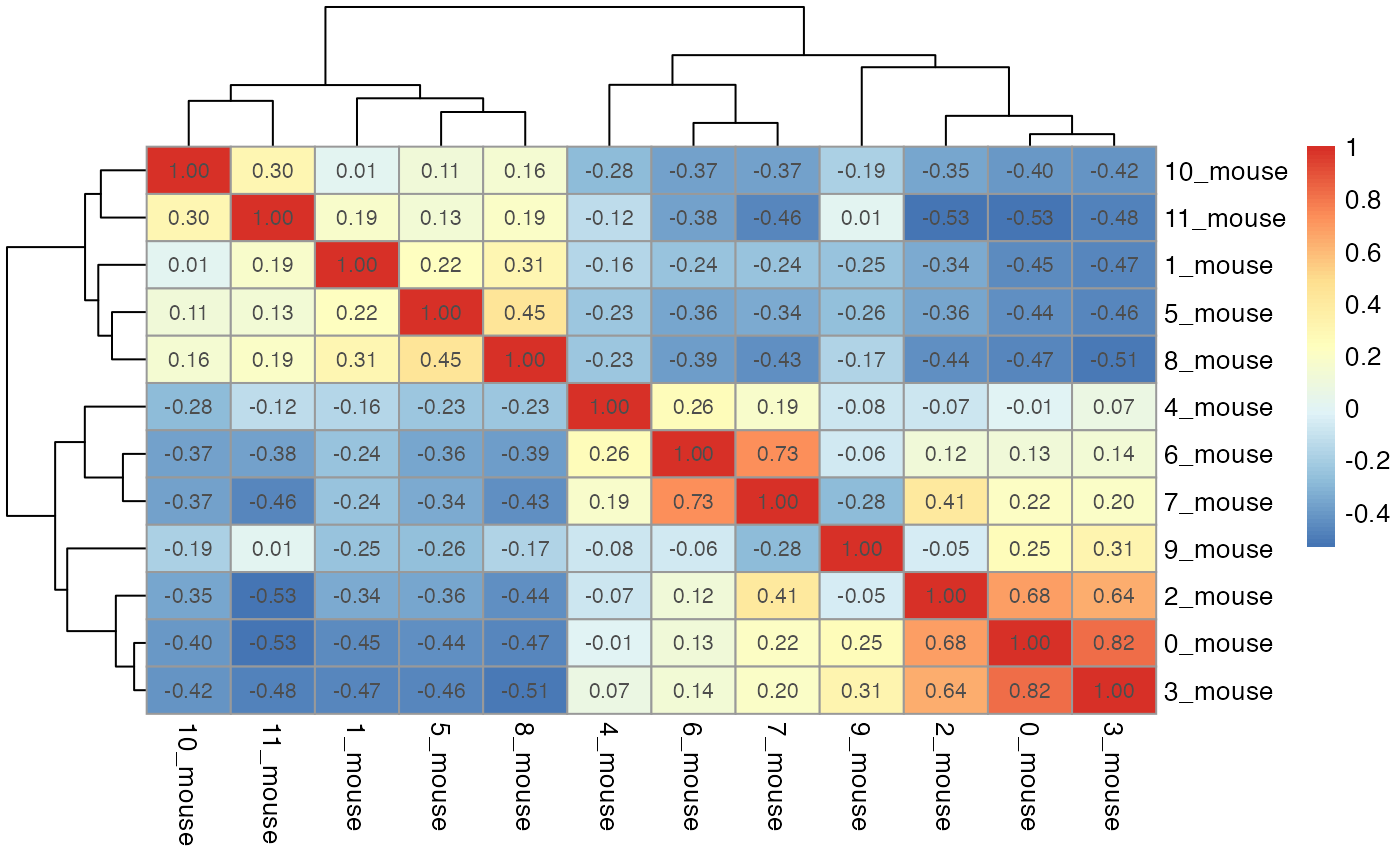
dist_coef <- getDistMat(seu_list, downsampling.size = 50)
#>
|
| | 0%
|
|=================================== | 50%
|
|======================================================================| 100%
par(mfrow = c(length(seu_list),1))
for(i in which(sapply(dist_coef, function(x) return(!is.null(x))))){
tmp <- dist_coef[[i]] + t(dist_coef[[i]])
diag(tmp) <- 1
pheatmap::pheatmap(tmp, display_numbers = TRUE)
}

for(seu_itor in 1:2){
tmp <- dist_coef[[seu_itor]] + t(dist_coef[[seu_itor]])
diag(tmp) <- 1
tmp <- 1 - tmp
hc <- hclust(as.dist(tmp), method = "average")
hres <- cutree(hc, h = 0.4)
df_hres <- data.frame(hres)
df_hres$hres <- paste0(df_hres$hres, "_", unique(seu_list[[seu_itor]]$Batch))
seu_list[[seu_itor]]$inicluster_tmp <- paste0(seu_list[[seu_itor]]$seurat_clusters, "_", seu_list[[seu_itor]]$Batch)
seu_list[[seu_itor]]$inicluster <- df_hres$hres[match(seu_list[[seu_itor]]$inicluster_tmp,rownames(df_hres))]
}
# plot(as.dendrogram(hc), horiz = T)
p1 <- scatterPlot(seu_list[[1]], "tsne", "inicluster")
p2 <- scatterPlot(seu_list[[2]], "tsne", "inicluster")
plot_grid(p1,p2)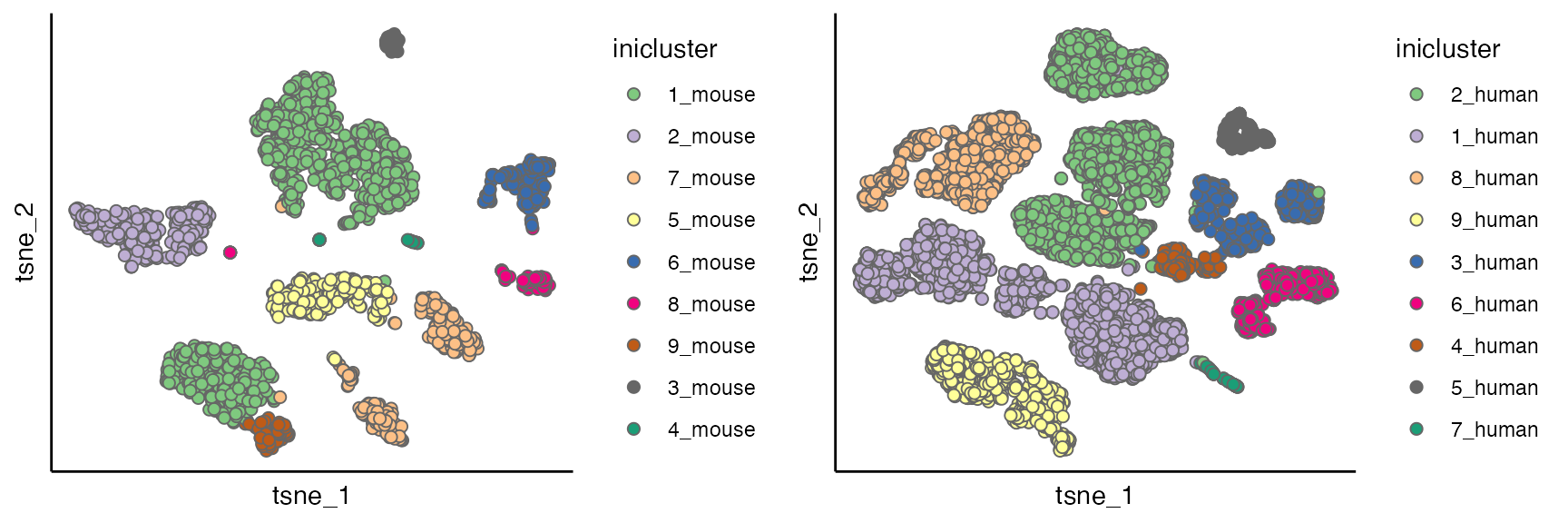
scatterPlot(seu_list[[2]], "tsne", "Group")
Calculate of IDER similarity matrix
res <- unlist(lapply(seu_list, function(x) return(x$inicluster)))
res_names <- unlist(lapply(seu_list, function(x) return(colnames(x))))
seu$initial_cluster <- res[match(colnames(seu), res_names)]
ider <- getIDEr(seu,
group.by.var = "initial_cluster",
batch.by.var = "Batch",
downsampling.size = 35,
use.parallel = FALSE, verbose = FALSE)
net <- plotNetwork(seu, ider, colour.by = "Group" , vertex.size = 0.6, weight.factor = 5)
hc <- hclust(as.dist(1-(ider[[1]] + t(ider[[1]])))/2)
plot(as.dendrogram(hc), horiz = TRUE)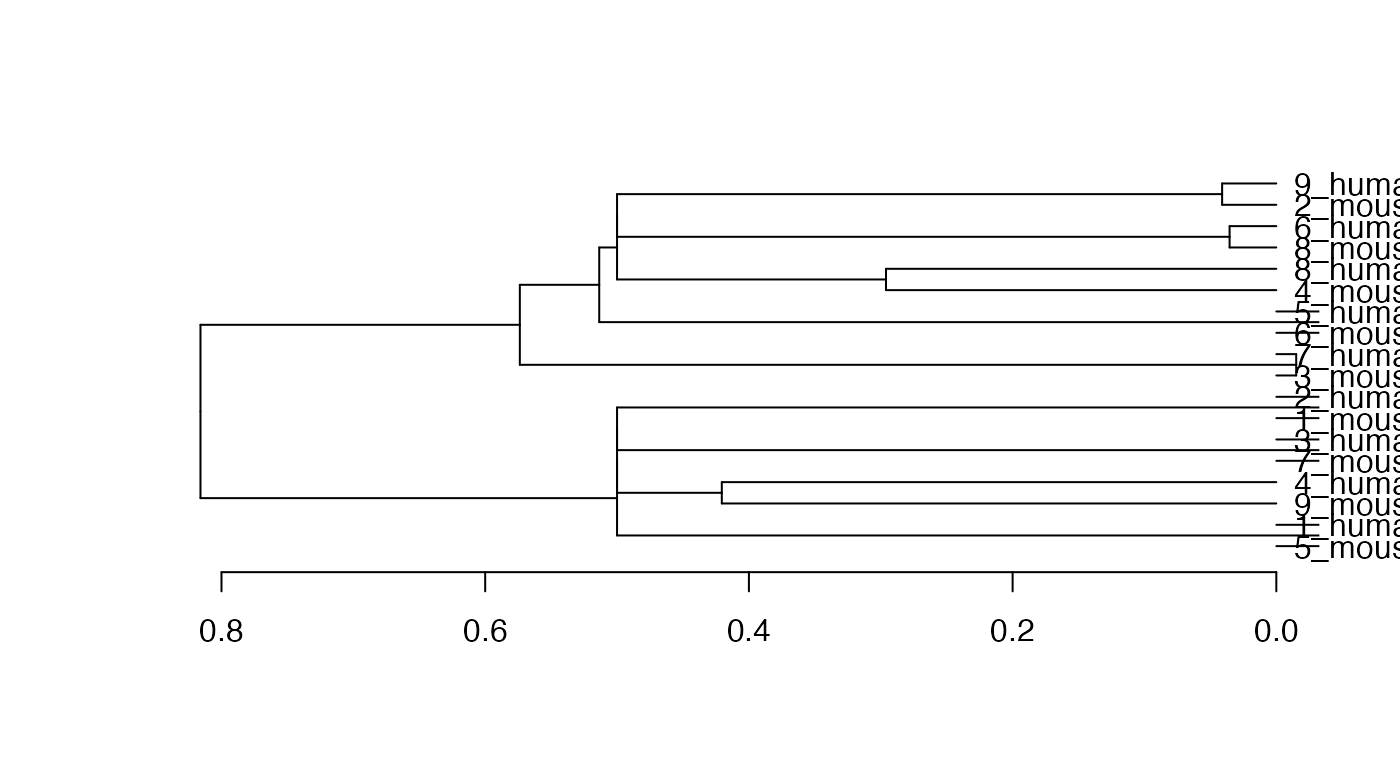
Perform final Clustering
seu <- finalClustering(seu, ider, cutree.h = 0.35) # final clustering
seu <- NormalizeData(seu, verbose = FALSE)
seu <- FindVariableFeatures(seu, selection.method = "vst",
nfeatures = 2000, verbose = FALSE)
seu <- ScaleData(seu, verbose = FALSE)
seu <- RunPCA(seu, npcs = 20, verbose = FALSE)
seu <- RunTSNE(seu, reduction = "pca", dims = 1:12)
plot_list <- list()
plot_list[[1]] <- scatterPlot(seu, "tsne", colour.by = "CIDER_cluster", title = "asCIDER clustering results")
plot_list[[2]] <- scatterPlot(seu, "tsne", colour.by = "Group", title = "Ground truth of cell populations")
plot_grid(plotlist = plot_list, ncol = 2)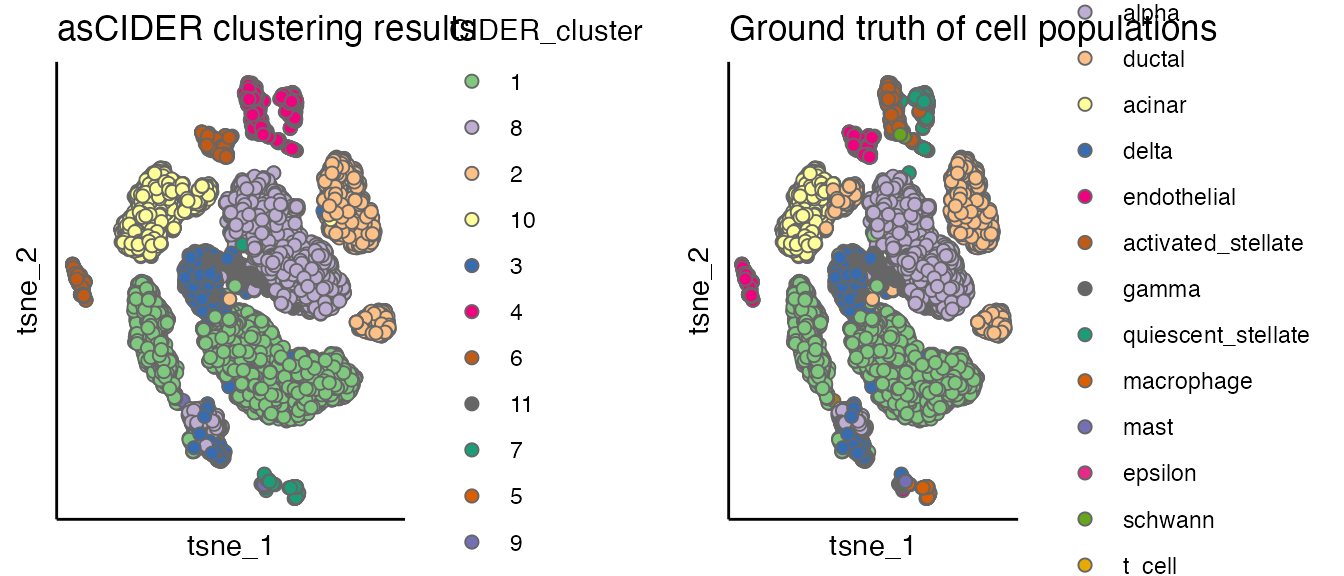
Technical
sessionInfo()
#> R version 4.1.2 (2021-11-01)
#> Platform: x86_64-apple-darwin17.0 (64-bit)
#> Running under: macOS Big Sur 10.16
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRlapack.dylib
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> attached base packages:
#> [1] parallel stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] cowplot_1.1.1 SeuratObject_4.0.4 Seurat_4.1.0 CIDER_0.99.1
#>
#> loaded via a namespace (and not attached):
#> [1] systemfonts_1.0.2 plyr_1.8.6 igraph_1.2.8
#> [4] lazyeval_0.2.2 splines_4.1.2 listenv_0.8.0
#> [7] scattermore_0.7 ggplot2_3.4.2 digest_0.6.28
#> [10] foreach_1.5.1 htmltools_0.5.2 viridis_0.6.2
#> [13] fansi_0.5.0 magrittr_2.0.1 memoise_2.0.0
#> [16] tensor_1.5 cluster_2.1.2 doParallel_1.0.16
#> [19] ROCR_1.0-11 limma_3.50.0 globals_0.16.1
#> [22] matrixStats_0.61.0 pkgdown_2.0.7 spatstat.sparse_2.0-0
#> [25] colorspace_2.0-2 ggrepel_0.9.3 textshaping_0.3.6
#> [28] xfun_0.28 dplyr_1.1.2 crayon_1.5.2
#> [31] jsonlite_1.7.2 spatstat.data_2.1-0 survival_3.2-13
#> [34] zoo_1.8-9 iterators_1.0.13 glue_1.6.2
#> [37] polyclip_1.10-0 gtable_0.3.0 leiden_0.3.9
#> [40] kernlab_0.9-29 future.apply_1.8.1 abind_1.4-5
#> [43] scales_1.2.1 pheatmap_1.0.12 DBI_1.1.1
#> [46] edgeR_3.36.0 miniUI_0.1.1.1 Rcpp_1.0.7
#> [49] viridisLite_0.4.0 xtable_1.8-4 reticulate_1.22
#> [52] spatstat.core_2.3-1 htmlwidgets_1.5.4 httr_1.4.2
#> [55] RColorBrewer_1.1-2 ellipsis_0.3.2 ica_1.0-2
#> [58] farver_2.1.0 pkgconfig_2.0.3 sass_0.4.0
#> [61] uwot_0.1.10 deldir_1.0-6 locfit_1.5-9.4
#> [64] utf8_1.2.2 labeling_0.4.2 tidyselect_1.2.0
#> [67] rlang_1.1.1 reshape2_1.4.4 later_1.3.0
#> [70] munsell_0.5.0 tools_4.1.2 cachem_1.0.6
#> [73] cli_3.4.1 dbscan_1.1-8 generics_0.1.1
#> [76] ggridges_0.5.3 evaluate_0.14 stringr_1.5.0
#> [79] fastmap_1.1.0 yaml_2.2.1 ragg_1.1.3
#> [82] goftest_1.2-3 knitr_1.36 fs_1.5.0
#> [85] fitdistrplus_1.1-6 purrr_1.0.1 RANN_2.6.1
#> [88] pbapply_1.5-0 future_1.28.0 nlme_3.1-153
#> [91] mime_0.12 compiler_4.1.2 rstudioapi_0.13
#> [94] plotly_4.10.0 png_0.1-7 spatstat.utils_2.2-0
#> [97] tibble_3.2.1 bslib_0.3.1 stringi_1.7.5
#> [100] highr_0.9 desc_1.4.0 lattice_0.20-45
#> [103] Matrix_1.3-4 vctrs_0.6.2 pillar_1.9.0
#> [106] lifecycle_1.0.3 spatstat.geom_2.4-0 lmtest_0.9-39
#> [109] jquerylib_0.1.4 RcppAnnoy_0.0.19 data.table_1.14.2
#> [112] irlba_2.3.3 httpuv_1.6.3 patchwork_1.1.1
#> [115] R6_2.5.1 promises_1.2.0.1 KernSmooth_2.23-20
#> [118] gridExtra_2.3 parallelly_1.32.1 codetools_0.2-18
#> [121] MASS_7.3-54 rprojroot_2.0.2 withr_2.5.0
#> [124] sctransform_0.3.3 mgcv_1.8-38 grid_4.1.2
#> [127] rpart_4.1-15 tidyr_1.3.0 rmarkdown_2.11
#> [130] Rtsne_0.15 shiny_1.7.1